Abstract
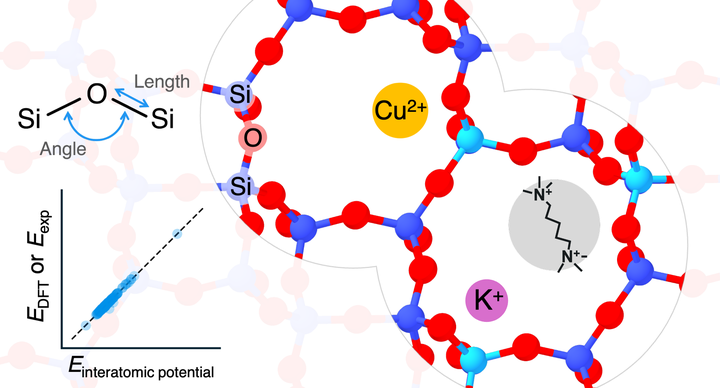
Interatomic potentials (IPs) with wide elemental coverage and high accuracy are powerful tools for high-throughput materials discovery. While the past few years witnessed the development of multiple new universal IPs that cover wide ranges of the periodic table, their applicability to target chemical systems should be carefully investigated. We benchmark several universal IPs using equilibrium zeolite structures as testbeds by evaluating geometric parameters, energies, and relative stability. We select a diverse set of universal IPs encompassing two major categories: (i) universal analytic IPs, including GFN-FF, UFF, and Dreiding; (ii) pretrained universal machine learning IPs (MLIPs), comprising CHGNet, ORB-v3, MatterSim, eSEN-30M-OAM, PFP-v7, and EquiformerV2-lE4-lF100-S2EFS-OC22. We compare them with established tailor-made IPs, SLC, ClayFF, and BSFF using experimental data and density functional theory (DFT) calculations with dispersion correction as the reference. The tested zeolite structures comprise pure silica frameworks and aluminosilicates containing copper species, potassium, and an organic cation. We found that GFN-FF is the best among the tested universal analytic IPs, but its good performance is limited in silica zeolites without highly strained rings. Some universal MLIPs achieve the energies with a root mean squared error between MLIP and DFT energies below a previously tailor-made MLIP. Among the universal MLIPs, the eSEN-30M-OAM model outperformed the other universal IPs in predicting structures and energies close to experiments and DFT across all zeolite structures studied. These findings show that the modern pretrained universal MLIPs are practical tools in replacing high-throughput DFT calculations against equilibrium zeolite structures, although they inherit the inherent errors of DFT.