Density Functional Theory Study of Deoxydehydration Reaction by TiO2-Supported Monomeric and Dimeric Molybdenum Oxide Catalysts
概要
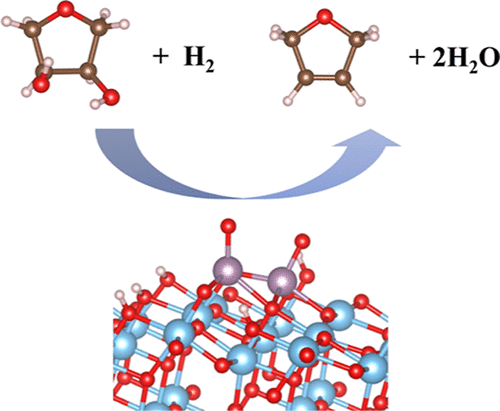
The development of efficient heterogeneous catalysts for converting biomass into value-added chemical compounds remains at the forefront of catalysis research. Deoxydehydration (DODH) reaction that can transform vicinal hydroxy groups with the cis-configuration to the corresponding C═C bond in a single step is one of the promising techniques, and molybdenum oxide catalysts supported on TiO$_2$ have been reported as an effective catalyst using hydrogen as a reducing agent. Here, using density functional theory calculations, structures of monomeric and dimeric molybdenum oxide catalysts supported on anatase TiO$_2$(101) have been determined, and we decipher the reaction mechanisms of the conversion of 1,4-anhydroerythritol to 2,5-dihydrofuran over these catalysts as a model reaction. We have found that MoO$_3$ and Mo$_2$O$_5$ are the most stable structures for monomeric and dimeric species that exhibit the oxidation states of MoVI and MoV–MoVI, respectively, under the experimental conditions. For monomeric species, it is rather difficult to catalyze DODH reaction due to the instability for MoIV species and also the higher barrier for the C–O bond scission for MoV or MoVI species. For dimeric species, structures with the oxidation state of MoIV–MoV or MoV–MoV that is found in the form of Mo$_2$O$_4$ exhibit promising energy profiles in terms of stability and energy barrier (∼1.0 eV) for the C–O bond dissociation. Considering the experimental facts that MoIV species is responsible for the DODH reaction and Mo–Mo bond is present, the MoIV–MoV structure could be the plausible active species. Our findings would provide useful information for the catalyst design using earth-abundant and less-expensive metal-based catalysts for the DODH reaction.